
Model potential for the description of metal/organic interface states
N. Armbrust, F. Schiller, J. Güdde, U. Höfer
Scientific Reports 7 (2017) 46561
An analytical one-dimensional model potential predicts the energy position and the wave function overlap of metal/organic interface states. It explains how the energy of the interface state depends systematically on the bond distance between the carbon backbone of the adlayers and the metal. Comparison with numerous experimental results demonstrates the general applicability and robustness of the model.
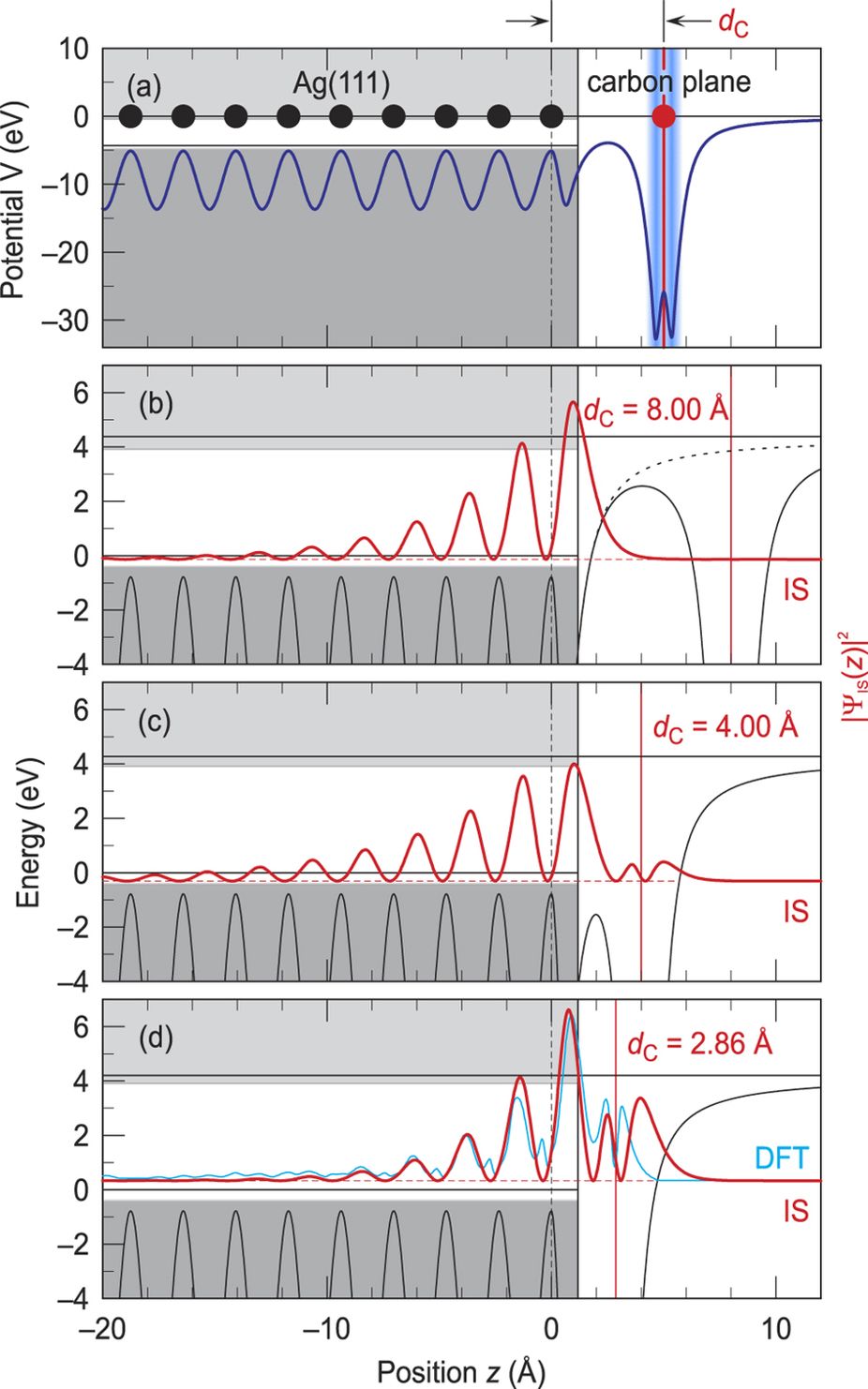
One-dimensional model potential V(z) for a carbon layer on Ag(111) and the probability densities |ΨIS(z)|2 of the interface state formed at differing metal-C distances.
Charge transfer at the interface between a metal and a layer of organic molecules plays a decisive role in the functionality of organic semiconductor devices and for future applications of molecular electronics. It depends crucially on the energy alignment and the wave function overlap of electronic states at such interfaces. Of particular interest for model studies are interface-specific electronic states like the ones observed for PTCDA and other π-conjugated organic molecular layers grown on Ag(111). These interface states originate from the Shockley surface state of the bare metal substrate. This state is upshifted due to the interaction with the molecular layer while hybridization of molecular and metallic states remains rather small in the region of the projected band gap of the metal.
In order to highlight the main physical mechanism for the formation of the delocalized interface state at organic/metal interfaces without the help of complex DFT calculations, Armbrust and coworkers propose a one-dimensional description by an analytical model potential. The choice of the potential is inspired by previous work on surface states of clean metals. It utilizes graphene as a universal representation of flat-lying adlayers of π-conjugated organic molecules. Unlike other, more adsorbate-specific model potentials used previously to describe interfacial electronic states, its main parameter is simply the distance of the carbon plane from the metal substrate.
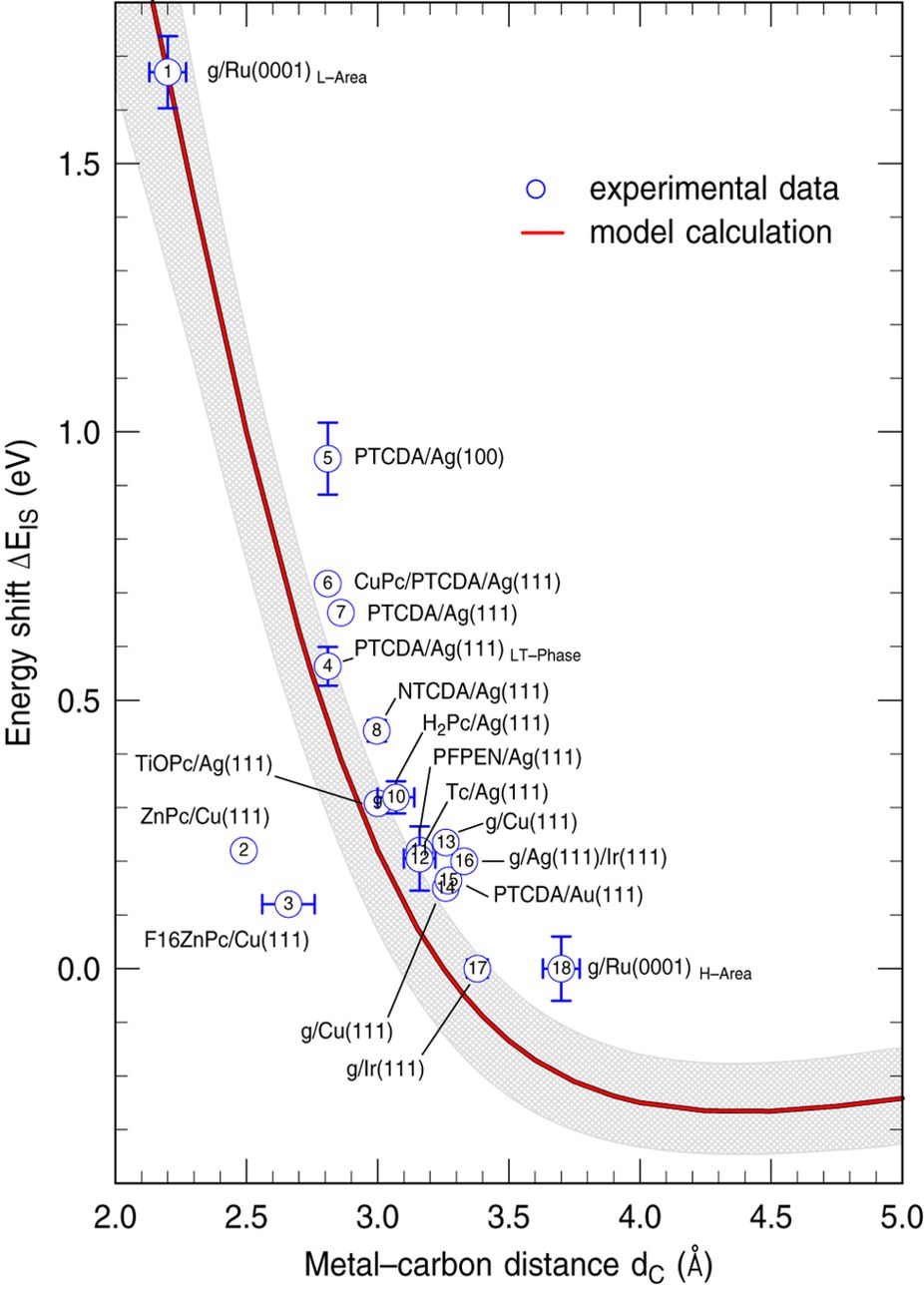
Energy shift of the interface state with respect to the energy of the former surface state on the bare metal as a function of the carbon-metal distance.
The work shows that the same model potential not only predicts the energy of the interface state in various graphene/metal systems, but can be applied to a large class of flat-lying molecular layers with a similar π-π interaction as in graphene. The model calculation clearly illustrates how the interface state develops from the former Shockley-type surface state of the bare metal substrate with increasing interaction between the molecular film and the metal. By comparing the results with available experimental data for different organic molecules, the authors show that their model is able to describe the systematic dependence of the energy of the interface state on the bond distance between the carbon backbone and the metal with predictive power. Moreover, the model reveals how the wave function overlap of the interface state with both the bulk metal and the molecular overlayer depends on the carbon-metal distance.